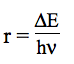
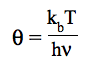
III.3 Description of the Calculation menu
KiSThelP has been designed to perform statistical mechanics calculations from ab initio quantum chemistry data. This results in the computation of canonical partition functions, molecular thermodynamic properties, thermal equilibrium constants, transition state theory (TST) rate coefficients, and RRKM rate coefficients. One-dimensional tunneling and variational effects have also been incorporated in the TST calculations.
It is to be noted that the standard state of pure gaseous species (ideal gas at P0=1 bar) is used in chemical equilibrium and TST kinetic properties computations, but the molecular properties calculated with KiSThelP can also be estimated at any pressure. These properties computations clearly have some limitations the user must be aware of. The Born-Oppenheimer and ideal gas approximations are invoked. The electronic partition function is truncated after the first term. Harmonic oscillator and the rigid-rotor approximation are assumed.
At a given temperature, the data required to derive the vibrational partition functions Qvibv=0 from ab initio calculations are the frequencies (νi) of vibrational modes obtained within the harmonic oscillator approximation. In calculating the vibrational partition function, vibrational energy is measured from the ground state (v=0). But, we also print out QvibBOT obtained by choosing the zero of energy to be the bottom of the potential energy curve. Due to the neglect of anharmonicity in electronic structure computations, it is known that there is a systematic error in the calculated harmonic vibrational frequencies when they are compared to the experimental fundamental vibrational frequencies. A scaling factor is commonly used to correct the calculated value to match the experimental one. KiSThelP gives the possibility to scale harmonic vibrational frequencies by such a factor (directly from the GUI). The harmonic oscillator approximation provides a fast and straightforward method to determine the vibrational frequencies used to compute the partition function of internal modes, but it breaks down for large-amplitude internal motions such as internal rotations. The hindered rotor approximation offers a significant improvement as compared to the harmonic oscillator treatment. That is the reason why the approach of Flagan et al [B. R. McClurg, W. A. Goddard and R. C. Flagan, J. Chem. Phys. 106 (1997) 6675] has been implemented in KiSThelP to account for deviations from the harmonic oscillator (HO) value. The calculations are based on two quantities:
θ = kbT/hν and r = ΔE/hν. The deviations proposed by the authors for the vibrational contributions are:
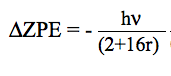
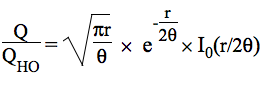
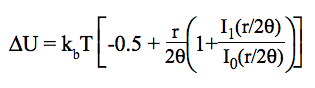
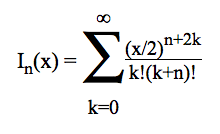
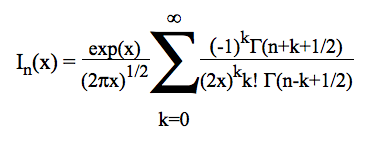
1) Atoms and Molecules :
This menu is to compute statistical and thermodynamical properties of a system (atom or molecule, or TS, or reactionpath point)
KiSThelP can compute
these properties :
- at a
single temperature
-
within a temperature range
- at a
single pressure
- within
a pressure range
The equations used for computing statistical and thermochemical data in KiSThelP are from standard texts on thermodynamics (see for example [R.A. Alberty and R. J. Silbey, "Physical Chemistry",1st edition, Edited by John Wiley & sons, Inc, ]. Formula employed in KiSThelP are here . The starting point in each case (translational, vibrational, rotational) is the partition function Q(V,T) for the corresponding component of the total partition function.
In addition to the numerical results (statistical and thermodynamical), the Maxwell-Boltzmann
distribution (Ni/N) = g(Ei)exp(-Ei/kBT)/Q is plotted as a function of (Ei/kBT). Ei represents energy levels in the translational and ro-vibrational modes. In practice, the degeneracy g(Ei) is evaluated as the product N(Ei) × dEi using the density of states and a small energy grain dEi, respectively.
N(Ei) is obtained by convolution using ro_vibrational and translational densities of states (as described by W. Forst [in "Theory of Unimolecular reactions", Academic Press, New York, 1973 and "Unimolecular reactions" , Cambridge University Press,2003]. It also includes electronic degeneracy of the ground state. The density of states of vibrations and rotations have been calculated using the Laplace-transform method, which is based on the inversion of the partition function. For translational modes, a semi-classical approach is employed here, rather than the Laplace-transform method.
KiSThelP is an interactive program that allows the user to change input data directly through the GUI. For single molecular system calculations, the user can interactively test the influence of the mass, potential energy, electronic degeneracy, rotational symetry number, inertia moment, vibrational frequencies, on statistical and thermodynamical properties. The "default values" button restores initial values. Also, a "vibrational" scaling factor field is made available. Default value is 1.0, meaning that no scaling factor is applied. Changing to values ≤ 1 will multiply each initial frequency by this factor.
Similarly, temperature and pressure can be interactively changed from single value to range of values. All the properties are immediately re-computed accounting for new parameters. In the case of a "temperature range" or "pressure range", a 2D-plot is displayed. Selected properties can then be displayed individually.
This grahical tools offers several facilities: scales settings, changing titles x/y limits, exporting image (png format), saving numerical data …
An input file must be supplied (.kinp or data fetched from electronic structure calculation results).
2) k / TST :
Classical Transition State Theory (TS located at the first-order saddle point)
The method used is the conventional transition state theory [H. Eyring, Chemical Reviews, 17 (1935) 65]. For an overview of TST and its improvements, see also [A. Fernandez-Ramos, B. A. Ellingson, B. C. Garrett, and D. G. Truhlar, Chapter 3 in Reviews in Computational Chemistry, 23 (2007) 125 , edited by K. B. Lipkowitz and T. R. Cundari (Wiley-VCH, Hoboken, NJ)].
Conventional TST requires information only for the saddle point and reactant(s). The equation usually presented for the conventional TST is:
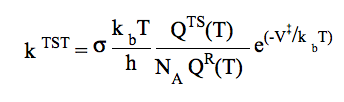
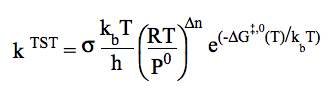
* UniMolecular
Reaction : to compute an unimolecular rate constant
at the Transition State Theory (TST) level
KiSThelP will ask the user for two data filemanes: one for the reactant, one for the TS (see ReactionPath section)
* BiMolecular Reaction : to compute a bimolecular rate constant
at the Transition State Theory (TST) level
Reactants data are obtained from
separated fragments electronic structure calculations.
KiSThelP will ask the user for three data filemanes: two for
the reactant
1 and 2, one for the TS (see ReactionPath section)
KiSThelP can compute rate constants and activation properties:
- at a single temperature
- within a temperature range
- at a single pressure
- within a pressure range
(it is to be noted that, though the TST rate constant is based on the standard state of pure gaseous species (ideal gas at P0=1 bar), non-standard activation properties can be displayed too).
Temperature and pressure can be interactively changed from single value to range of values. All the properties are immediately re-computed accounting for new parameters. In the case of a "temperature range" or "pressure range", a 2D-plot is displayed. Selected properties can then be displayed individually.
This grahical tools offers several facilities: scales settings, changing titles x/y limits, exporting image (png format), saving numerical data …
In addition to all the previous (numerical, graphical) results, the micro-canonical rate constant k(E) in s-1 for an unimolecular reaction is displayed as a function of E/kbT. This involves sum and density of states computations as presented further below in section 8 (RRKM).
KiSThelP also performs two statistical fits to the two- and three-parameters Arrhenius equations : k = Ae -Ea/RT and k = A × Tn e -Ea/RT. In the single temperature mode, teh Arrhenius fit is obtained from a temperature range centred on the selected temperature. In the range
temperature mode, the selected range serves to obtain the fit. The corresponding plots are compared to the TST k=f(T) curves. Numerical and graphical results can be saved through the plot tool.
3) k / TST / W :
Classical Transition State Theory (TS located at the first-order saddle point) including Wigner tunneling correction
One-dimensional quantum mechanical effects on reaction coordinate motion (tunneling and non-classical reflections) are incorporated by a multiplicative transmission coefficient χ(T), so that: kTST/T(T) = χ(T) × kTST(T). In this relative simple treatment, the imaginary frequency (characterizing the reaction mode) is used to compute the Wigner tunneling correction [E. Wigner, Z. Phys. Chem. B, 19 (1932) 203].
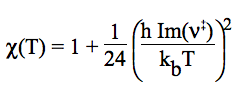
4) k / TST / Eck :
Classical Transition State Theory (TS located at the first-order saddle point) including Eckart tunneling correction
A Quantum effects in the reaction mode degree of freedom are
included by multiplying the TST rate constant by a transmission coefficient. One-dimensional quantum mechanical tunneling treatment through an unsymmetrical Eckart potential energy barrier is involved [C. Eckart, Phys. Rev. 35 (1930) 1303 ]. This methodology requires no ab initio calculations at points other than reactants, products, and saddle point. User will be asked for the ZPE corrected reverse energy barrier. The tunneling effect is accounted for by a multiplicative factor, so that: kTST/T(T) = χ(T) × kTST(T).
The analytic form proposed by Eckart can model a variety of physically reasonable shapes (involving unsymmetrical forms) and admits an analytical solution of the corresponding Schrödinger equation and then for the probability p(E) of transmission through the corresponding 1-D barrier at energy E. Original formula are used in KiSThelP with L, the width of the transition region which can be defined in term of the imaginary frequency ν‡ characterizing the reaction mode (that is routinely obtained from electronic structure calculations).
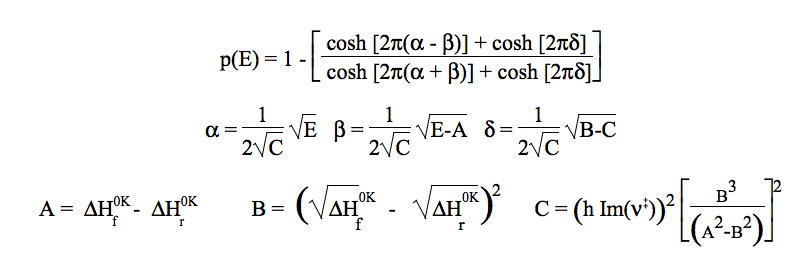
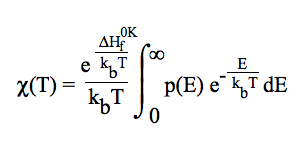
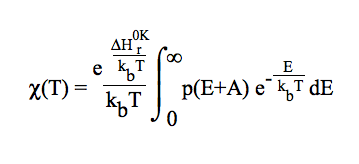
5) k / VTST :
Generalized Transition State Theory (TS located variationally; the optimum TS corresponds to a maximum free energy of activation at a given temperature)
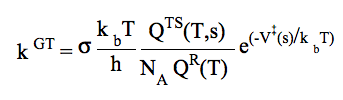
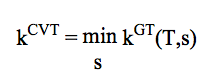
In VTST, the required information is the Minimum Energy Path (MEP) information obtained from electronic structure calculations. This information is read from a raectionpath .kinp input file containing as "POINT" sections as points calculated along the reaction path (including the transition state). The coordinate reaction, potential energy, inertia moment, ...
(see ReactionPath section) must be supplied for each point.Remarks:
- As abovementioned for conventional TST, the reaction path multiplicity is given by the user through KiSThelP graphical interface
- Along the reaction path, ideally, all bound modes transverse to the reaction path should be characterized by no imaginary frequency in KiSThelP. But practically, from electronic structure calculations, a reactionpath point can have 0, 1, or more than one imaginary boundary modes normal to the reaction mode. In KiSThelP, 0 or 1, or more imaginary frequencies will be accepted for a reaction path point. But if 0 imaginary frequency is found, then, to handle a homogeneous set of data the smallest frequency is set to imaginary number to design the reaction mode (thus it is removed from statistical and kinetics treatments). If more than 1 imaginary frequency are found, corresponding imaginary frequencies are removed from statistical treatments. In all these cases, warnings will be delivered to the user.
6) k / VTST / W :
Generalized Transition State Theory (TS located variationally; the optimum TS corresponds to a maximum free energy of activation at a given temperature) including Wigner tunneling correction
To include quantum effects on reaction coordinate motion the rate constant kCVT(T) is multiplied by a transmission coefficient χ(T): kCVT/T(T) = χ(T) × kCVT(T). The imaginary frequency of the transition state (1-order saddle point on the potential energy surface) is used to compute the Wigner tunneling correction.7) k / VTST / Eck :
Generalized Transition State Theory (TS located variationally; the optimum TS corresponds to a maximum free energy of activation at a given temperature) including Eckart tunneling correction
The tunneling effect is accounted for by a multiplicative factor, so that: kCVT/T(T) = χ(T) × kCVT(T). A one-dimensional quantum mechanical tunneling treatment through an unsymmetrical Eckart potential energy barrier is involved. This methodology requires no ab initio calculations at points other than reactants, products, and saddle point. User will be asked for the ZPE corrected reverse energy barrier.
8) RRKM:
Treatment of unimolecular processus in gas phase. Transition state theory reformulated on a microcanonical basis. Pressure effects can be accounted for.
- KiSThelP will ask the user for two data filemanes: one for the
reactant, one for the TS (see ReactionPath section)
- Next, KiSThelP will ask for molecular
mass (diluent gas), ε/kb (in K) and σ(cm) for reactant and diluent gas respectively. These data will serve to estimate the deactivation rate constant within the RRKM unimolecular kinectic mechanism. Data may be found using the KiSThelP "Data/Lennard-Jones parameters" menu
- The user can then interactively test the influence of the statistical factor, collision
efficiency and critical energy E0 values, on the unimolecular rate constant, through the GUI.
(* LooseTS : not implemented in this version)
The implementation of this method in KiSThelP is based on the following books: (1)[K.A. Holbrook, M.J. Pilling and S. H. Robertson, "Unimolecular Reactions", Wiley & sons Eds, 1996] (2)[P. J. Robinson and K.A. Holbrook,"Unimolecular Reactions" Wiley-Interscience, London, 1972], [W. Forst, "Unimolecular reactions" , Cambridge University Press,2003]. Only reaction paths with a well defined 1st-order saddle point on the potential energy surface are considered here (Tight TS). The other cases (Loose transition state) represent one of the perpectives for futur developpments of KiSThelP.
* TightTS : to compute an unimolecular rate constant using the RRKM theory within
the rigid TS model
- KiSThelP can compute the rate constant:
- at a single temperature
- within a temperature range<
- at a single pressure
- within a pressure range
The thermal rate constants of the unimolecular reactions
are obtained by applying the RRKM theory to predict
the falloff behaviour. The microcanonical rate constants k(E) are calculated by the standard
RRKM expression:
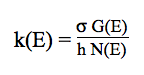
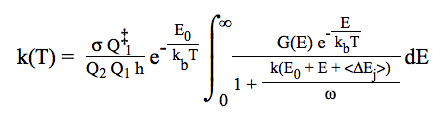
Calculation of gas-phase chemical reaction equilibrium constants
The knowledge of equilibrium constants is important in the
study of chemical reaction mechanisms. In addition to allowing
the prediction of the composition of a mixture, the equilibrium
constant is also connected to rate constants through
the existence of equilibrium between stable species in multiplestep
processes. Furthermore, in simulations of gas-phase mixtures,
backward rate can be found using the forward rate and the equilibrium constant.
In other respects, the equilibrium constant is a practical indicator
to test theoretical methods because it is very sensitive both to
the energy quality and to geometrical parameters and also
because the electronic structure of each compound that takes
part in the equilibrium is extremely different from the others.
To compute an equilibrium constant betwen reactant(s) and product(s)for a given balanced equation,stoichiometric coefficients must be supplied. The thermodynamical expression of the equilibrium constant for gas-phase reactions is employed in KiSThelP:
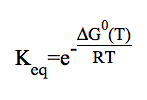
KiSThelP can compute gas-phase equilibrium constants
:
- at a
single Temperature
-
within a temperature range
10) Reset :
This menu will reset the current
calculation.