KiSThelP website
KiSThelP - a program to predict thermodynamic properties and rate constants from quantum chemistry results.
Screenshots
-- Screenshot 1 --
-- Screenshot 6 --
-- Screenshot 2 --
-- Screenshot 7 --
-- Screenshot 3 --
-- Screenshot 8 --
-- Screenshot 4 --
-- Screenshot 9 --
-- Screenshot 5--
-- Screenshot 10 --
-- Screenshot 11--
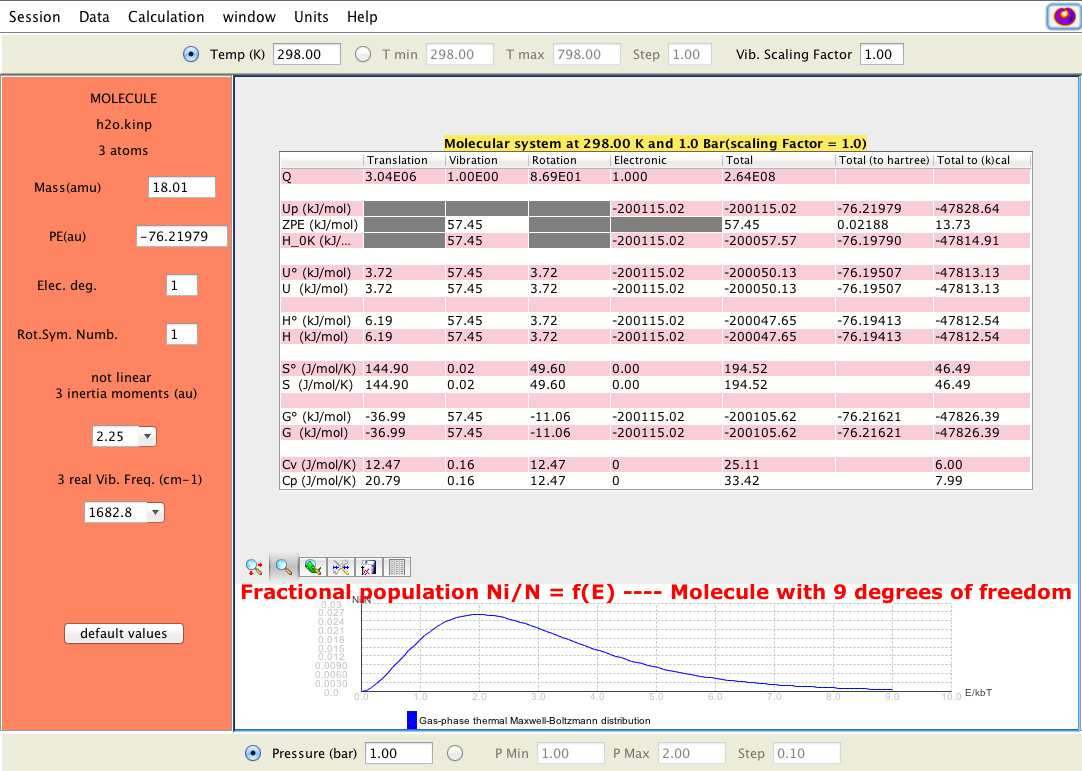
Output produced by KiSThelP showing statistical and thermodynamic properties of a molecule obtained from electronic structure data; temperature and pressure parameter fields are visible at the top and bottom; electronic structure data can be change in fields on left side.
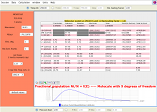
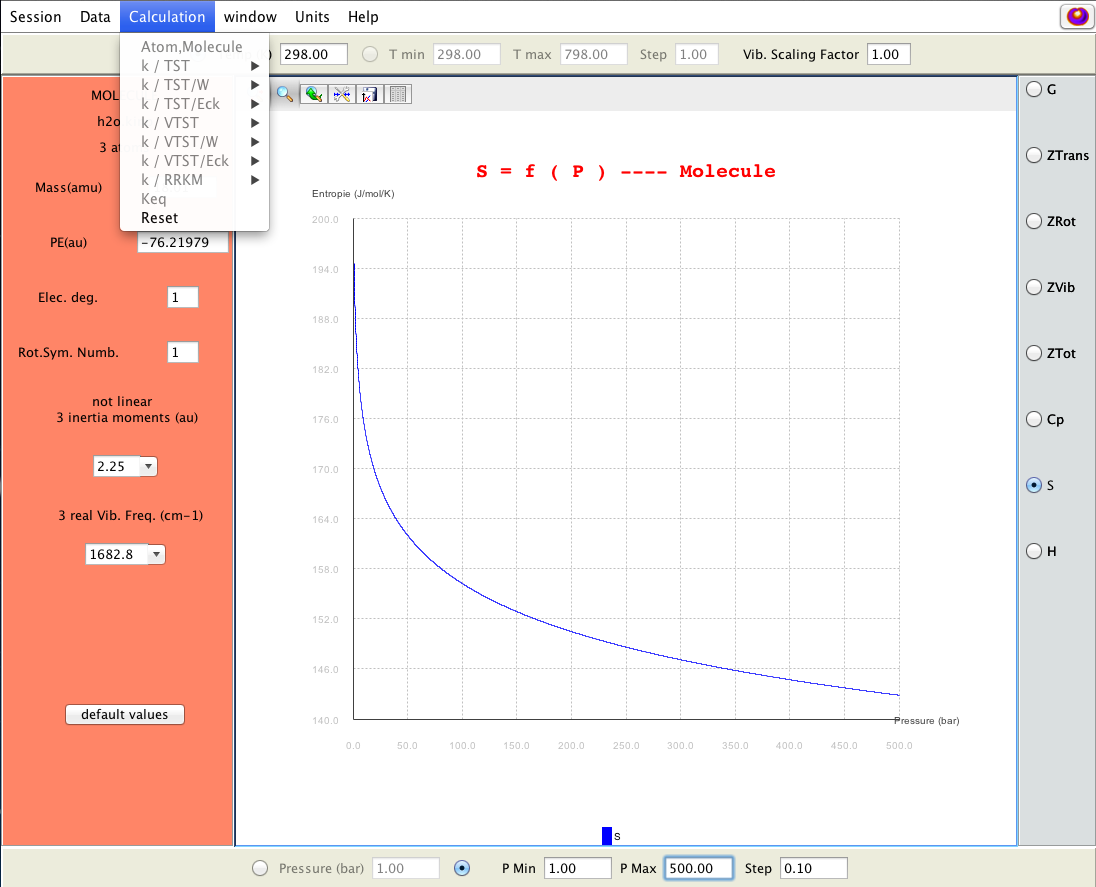
Molecular statistical and thermodynamic calculations; 2D-plot can be displayed for selected properties (buttons on right side); KiSThelP enables to change input data and temperature and pressure parameters directly through the GUI and to visually probe how it affects results.
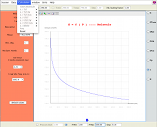
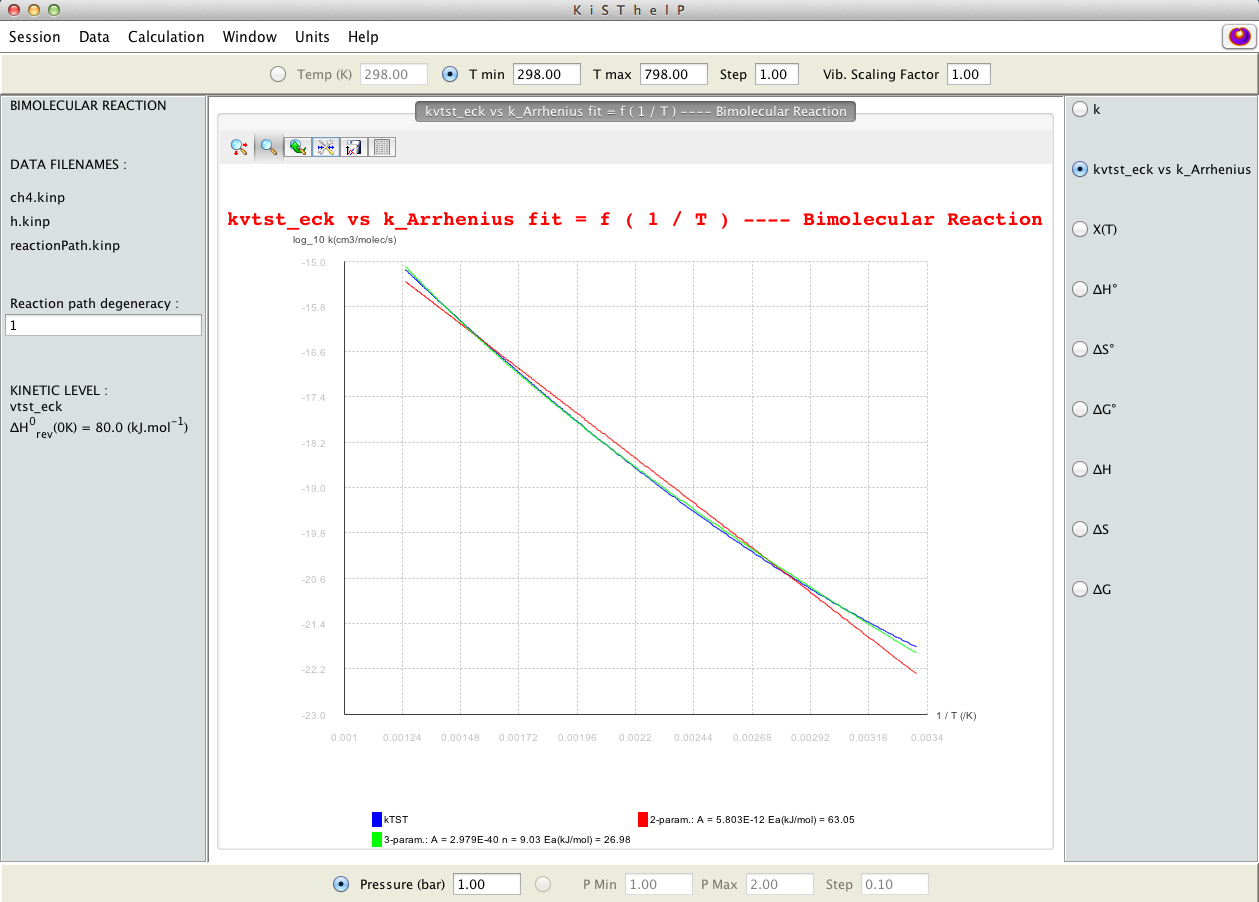
Logarithmic representation of rate constants against inverse temperature; prediction at four levels of theory (TST, VTST, TST/Tunnelling, VTST/Tunnelling); statistical fittings can be accessed from Arrhenius button (on right side); graph customization is possible (x and y ranges, …)
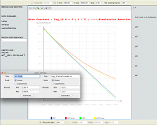
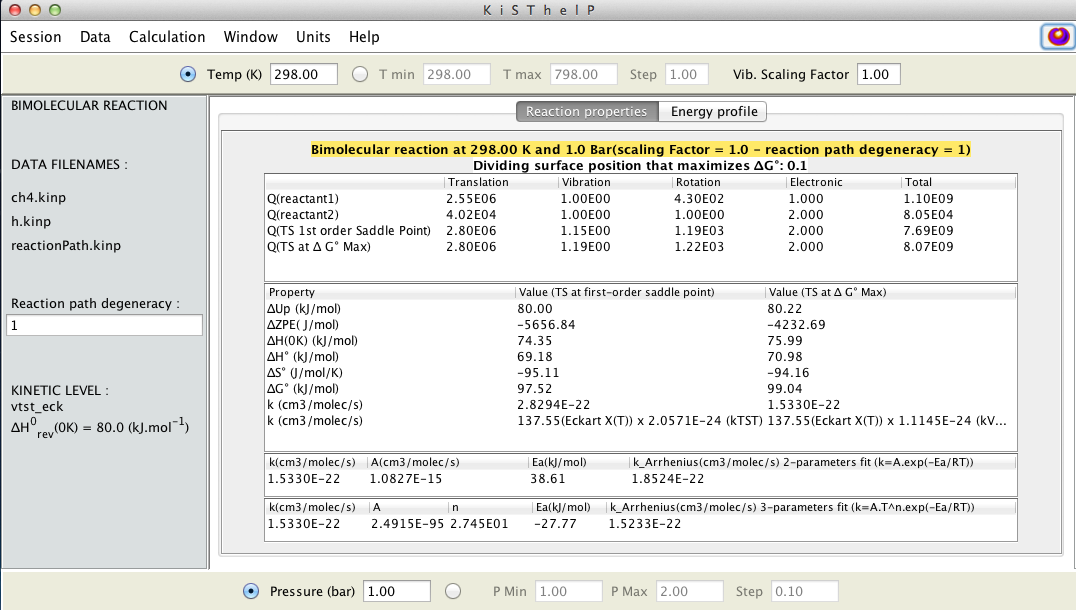
Full report for reacting species as well as TST kinetic results.
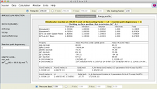
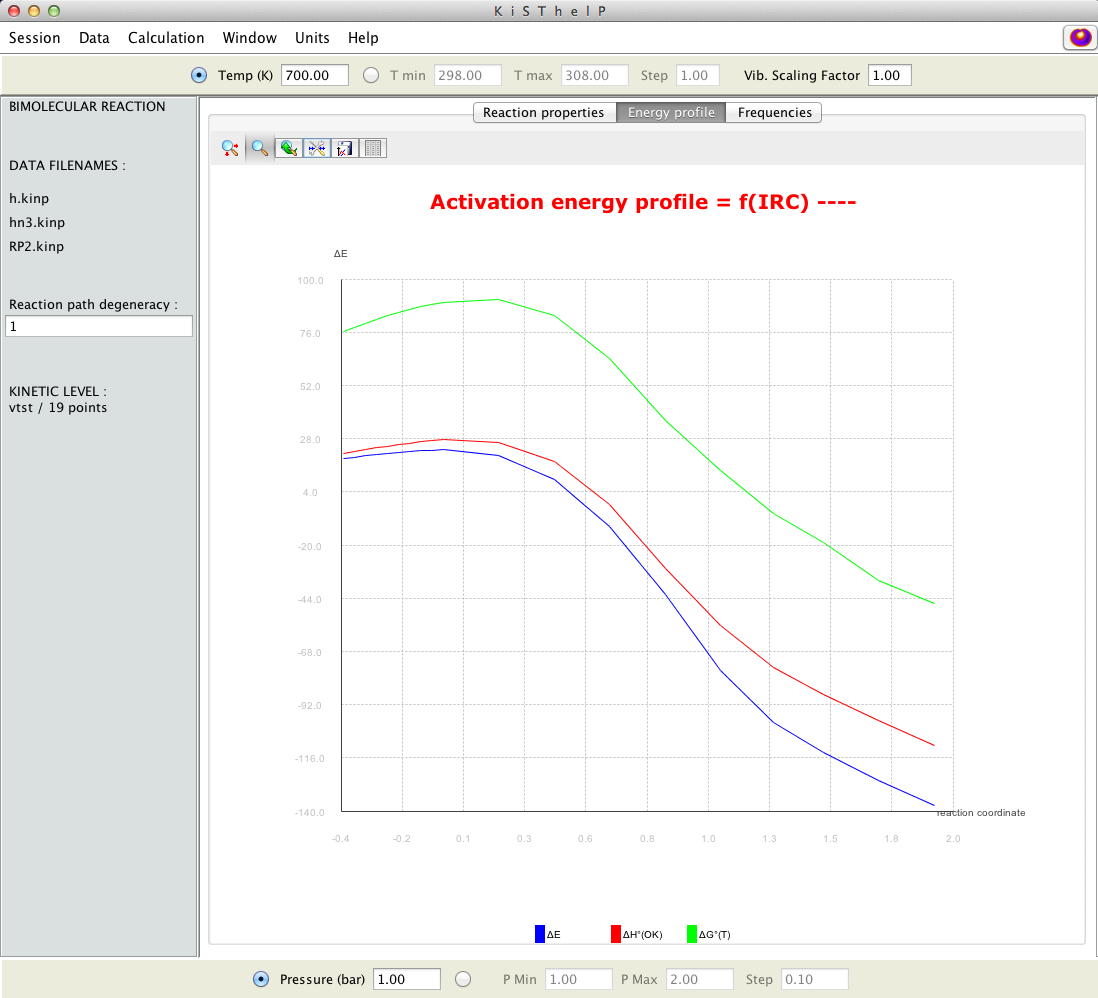
When a VTST calculation is invoked, a comparison between potential energy profile and the free energy profile is shown.
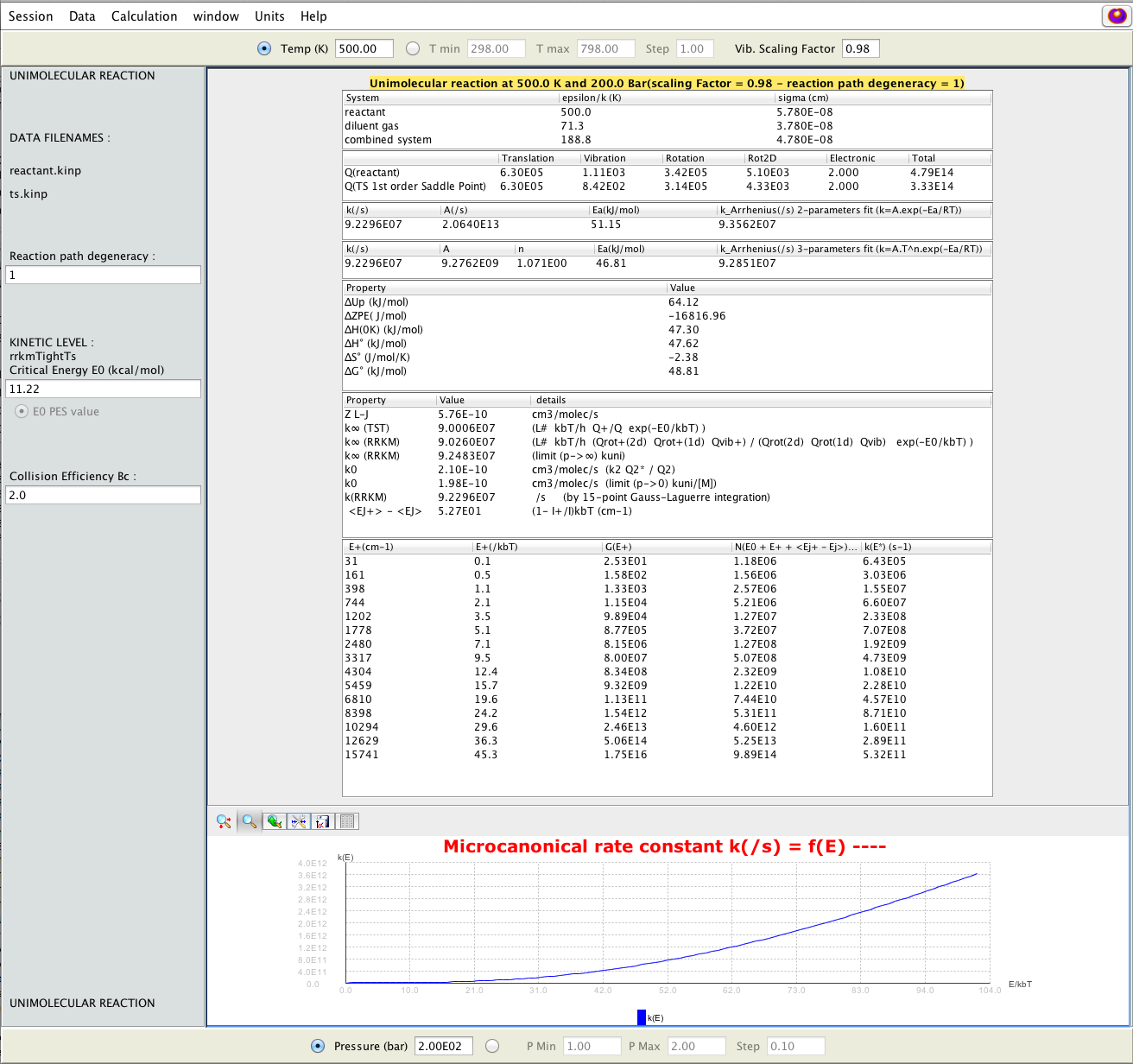
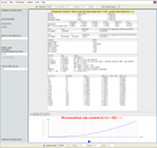
Full report of a RRKM simulation; data and parameters that were used to run the calculation are displayed (Lennard-Jones parameters, partition functions, thermal rate constants, high- and low-pressure limits, reaction properties, micro-canonical rate constants); energy threshold E0, collision efficiency, reaction path degeneracy can be changed interactively (left panel).
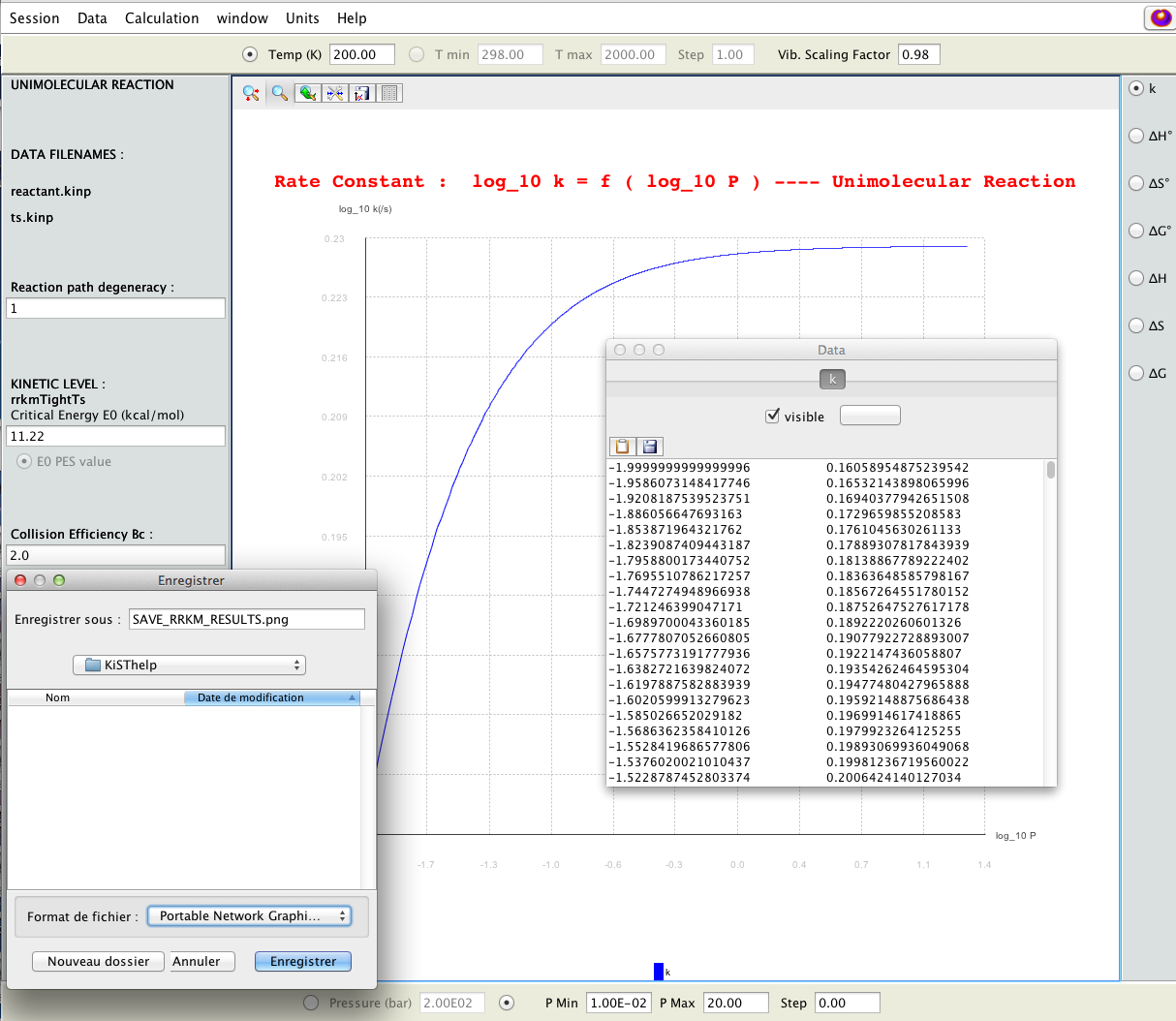
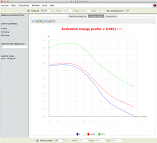
2D-plot of the RRKM falloff behaviour interactively obtained as a function of pressure P (bottom panel); numerical results as well as image can be saved on files.
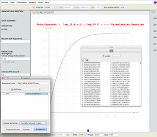
In order to perform a KiSThelP calculation, the user must prepare an ASCII input file (with file extension kinp). But alternatively, KiSThelP can read the needed electronic structure data directly from a computational chemistry software output file (this is the easiest way).
Gaussian (g03 and g09 versions),
GAMESS (2012) and
NWChem (6.0) output files are supported.

Statistical fittings of the two- and three-parameters Arrhenius equations.
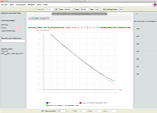
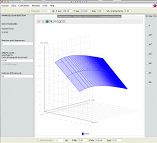
3D Plots of thermodynamics and kinetic properties as a function of temperature and pressure simultaneously.
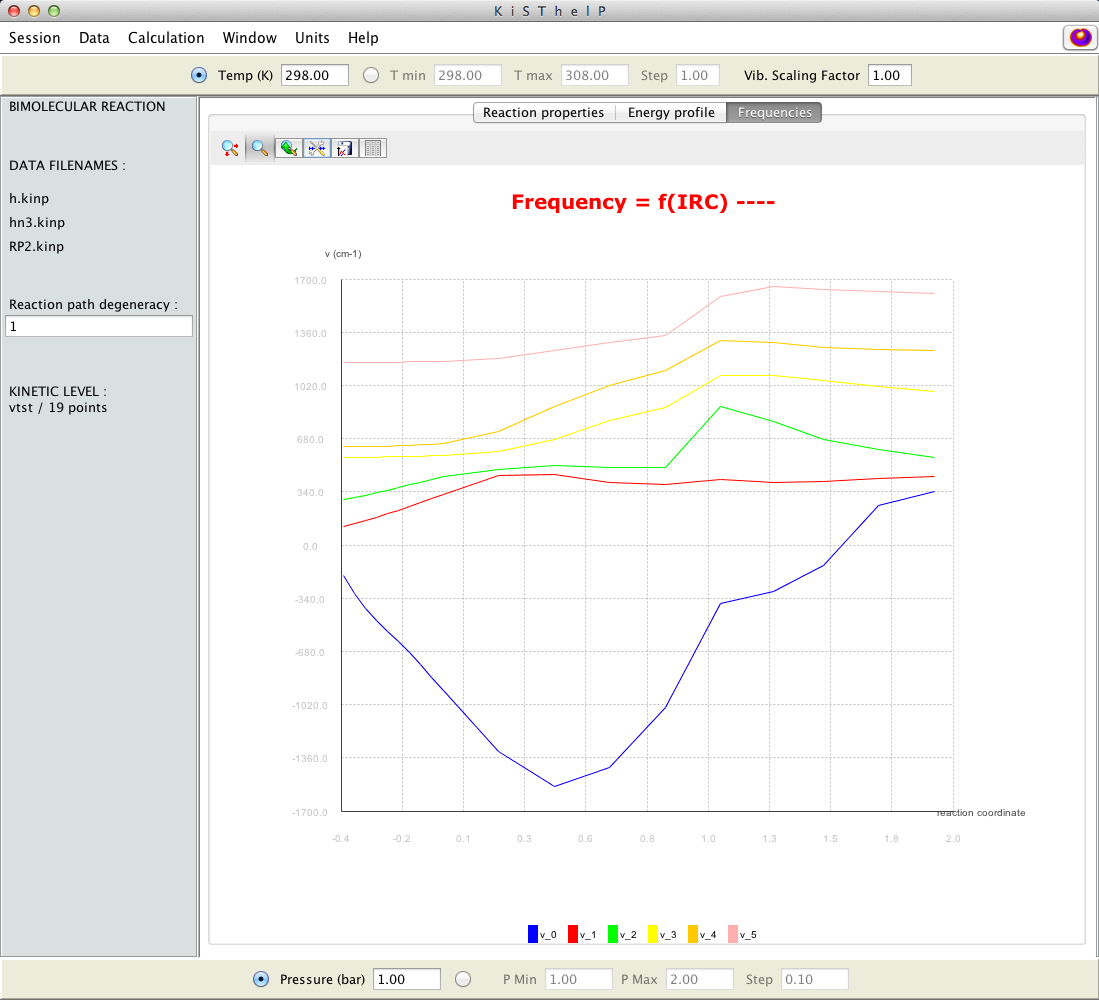
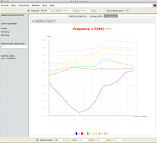
Vibrational frequencies along the IRC reaction path.
______________________________________________________________________________________________